テクノロジー
技術レポート:アーカイブ
Category:ライフサイエンス
大規模ゲノム解析パイプラインの構築
ゲノム診断技術は次世代ゲノムシークエンス手法の発展とともに急速に進化している。近い将来には全ゲノム解析(WGS:Whole Genome Sequencing)がゲノム診断の主流になり、医療施設において大規模なデータ解析が行われることが予想されることから、我々は高速かつ安定した動作の全ゲノム解析パイプラインの開発を進めている。本稿では、当社製品である「GenomeJack」に用いられている全ゲノムデータ解析技術について説明し、合わせて簡単な解析事例(がんゲノム解析)を紹介する。
参考情報:
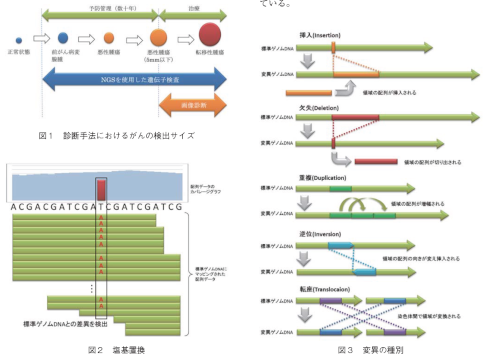
1 MSS技報・Vol.25 大規模ゲノム解析パイプラインの構築 Whole genome analysis pipeline, Parallelization and Visualization 野原 祥夫* 小原 康雄* 谷嶋 成樹* Sachio Nohara, Yasuo Ohara, Shigeki Tanishima ゲノム診断技術は次世代ゲノムシークエンス手法の発展とともに急速に進化している。近い将来には全ゲノム解析(WGS:Whole Genome Sequencing)がゲノム診断の主流になり、医療施設において大規模なデータ解析が行われることが予想されることから、我々は高速かつ安定した動作の全ゲノム解析パイプラインの開発を進めている。本稿では、当社製品である「GenomeJack」に用いられている全ゲノムデータ解析技術について説明し、合わせて簡単な解析事例(がんゲノム解析)を紹介する。 Genomic diagnosis has been rapidly developing with the advancement of next-generationsequencing. Among them, we focus attention on whole genome sequencing and data analysis,which would be the essential technology for genomic diagnosis in the near future. We explainwhole genome analysis for detecting cancer-related mutation and show the analysis case exampleusing our software GenomeJack. 1.まえがき 既に米国では23andMeなどが個人向けにDTC(注1)遺伝学的検査のサービスを展開しており、日本でも2014年にYahoo社やDeNA社などの大手IT企業が同分野に参入したことで、一般消費者にもゲノム解析や遺伝子検査に関する関心が高まっている。これらの遺伝学的検査は、変異が起こりやすいゲノム上の特定部位の塩基置換を判定するもので、自身の遺伝的背景や一般的な病気へのリスクなどを知ることができる。将来、検査結果と健康状態を結び付けるデータの蓄積が進めば、ビックデータ技術により健康管理ビジネス市場が大きく開けると言われている。 一方、医療機関における臨床応用の現場ではゲノム診断に利用可能な正確かつ多様なタイプの変異情報の抽出が求められている。次世代ゲノムシークエンサー(Next Generation Sequencing、以下「NGS」という)は、ヒトゲノムの構造を網羅的かつ正確に評価することが可能であり、さらに全遺伝子の発現量や遺伝子発現のON/OFFを制御しているゲノム修飾(エピゲノム)などの動的な状態の定量計測が可能なことから、がんの診断分野などで使用され始めている。現時点では、コストの問題で全ゲノム解析はあまり行われていないが、今後ともゲノムシーケンスコストが年率50%以下のペースで低下することは確実なため、数年のうちに全ゲノムデータ解析ががん診断の主流になると考えられている(注2)。 今回は当社製品のNGS向け高速ゲノム解析システムおよびゲノムブラウザであるGenomeJack Analysisの内部処理で用いられている全ゲノムデータ解析手法を紹介する。また、GenomeJackゲノムブラウザを用いてゲノムの変異を抽出する方法も紹介する。(注1) DTCとはDirect To Customerの略で直接消費者が利用できる。(注2) 現在、がん解析に必要なデータ量を用意するにはエクソーム解析では数十万円、全ゲノムデータ解析では数百万円の費用を要する。1. ゲノム解析技術⑴ がんゲノム解析の概要 がん検診では画像診断(PET、CT、MRIなど)が一般的に利用されている。がん治療においては兎に角早期発見が望まれるが、画像診断では5mm以下の微細ながんの検出が非常に困難である(図1)。NGSを使用したがんゲノム診断の場合は、末梢血中に分離されたがん細胞のDNA/RNAを検出することで、5mm以下の微細ながんも発見することができ[1]、前がん状態の検出が可能になる。さらに、NGSの特徴である定量的な計測により、がん細胞の変異割合を検出することができ、変異の種類によって効能が変化する抗がん剤(分子標的薬)*関西事業部 第五技術部2 MSS技報・Vol.25(図2)だけでなく、挿入・欠失、構造異常(重複・逆位・転座)、さらにコピー数異常(挿入・欠失・重複)など多岐に渡って検出する必要がある(図3)。エクソーム解析ではこれらすべての変異を検知することは不可能である。全ゲノム解析はこれらすべての変異を検出可能であるが、ゲノムシークエンスコストおよび大量のデータ解析を高速に行うための大規模データ解析技術が未完であることから、これまでは主流にはなりえなかった。ゲノムシーケンスコストの問題は解決されつつあり、データ解析技術が確立すれば、全ゲノム解析が主流になると考えられる。2. 当社の取り組み⑴ 全ゲノムデータ解析とは 全ゲノムデータ解析とは、抽出した細胞からヒトのゲノム配列(約30億塩基対)の全領域に対して解析を行う手法のことを言う。全ゲノムデータ解析は、取り扱うデータ量がテラバイト級であり、かつ、解析の基準となる標準ゲノム配列が特定の人種のものであるため、誤検出や検出漏れが多いという問題があり、解析時間の短縮と、検出感度、選択性の確保という面での改善が望まれている。の適切な選択を可能とし、治療効果を格段に高めることが可能である。 臨床応用分野でがん関連遺伝子の変異を抽出するために、患者から得られた正常細胞(これらは、生殖細胞系列:Germlineと呼ばれる)とがん細胞(体細胞系列:Somaticとよばれる)の比較解析により「がんドライバー変異」と呼ばれるがん化促進因子を抽出する。一般的に、がん組織として採取したサンプルの中には、複数種類のがん細胞(サブクローンと呼ばれる)が含まれる。サブクローンの型によりがんの性質が異なり、抗がん剤の効果も変わってくることから、サブクローン種類と比率の同定を確実に行える技術が望まれている[2]。⑵ なぜ全ゲノムデータ解析なのか 現在NGSに関連する解析手法は、その用途に合わせて多種多様になっている。塩基置換の検出方法の主流は遺伝子領域に限定して検出するエクソーム法である。これは解析する遺伝子領域を限定することで、低コスト化、高速化を実現している。しかし、がんの診断においてはサブクローンを正確に把握するためには、塩基置換図1 診断手法におけるがんの検出サイズ図2 塩基置換図3 変異の種別3 MSS技報・Vol.25ざまなRawデータフィルタ機能(クリップリード抽出、ロングメイトペアリード抽出など)を改良して高速データ表示を実現した。これらの改良により、変異の複雑な構造(図4)を理解するための表示多様性を実現している。⑵ 全ゲノムデータ解析のフロー(図5)[4] がん化は単一の遺伝子の変異のみに起因するのではな■解析時間 がんの診断などでは、ヒトゲノム全領域についてNGSを用い塩基配列を100倍(一般的にカバレージ、depthと言う)程度重複して読み込む必要がある。全ゲノムデータ解析でカバレージ100を実現するには、ヒトゲノムのサイズを30億塩基対とした場合には、約3,000億個の塩基情報を処理する必要がある。これを単一のサーバで解析するには1ヶ月以上の解析時間が必要となり、臨床現場で許容できる範囲を超えてしまう。この問題を解決するために、当社では解析ソフトウエアの処理を並列分散化し、100ノード以上のPCクラスタにて処理することで2日程度で答えを得ることが可能になっている。また、100ノード以上のPCクラスタを導入するには非常に高額なコストがかかるため、クラウドコンピューティングにより超並列解析環境を低コストで利用できるソフトウェアパッケージを開発した。■検出感度 ゲノム上に存在する反復配列など、NGSでは解読不能な領域が存在する。また、コピー数異常、構造異常は変異の構造が複雑であることが多く、現在普及している解析ソフトウエアでは十分な検出精度が得られないことが多い[3]。そのため、解析ソフトウェアの結果を統計的に再評価する二次処理プログラムの開発、複数の解析ソフトウェアの結果を並行して表示するブラウジング機能、変異検出に使用したRawデータと言えるマッピングデータとの関連性を容易に確認し、変異の証拠を明確化するブラウジング環境を開発した。ブラウジング環境は、高速なデータ表示を得意とするGenomeJackの改良により実現し、さらに、解析現場にて必要とされるさま図4 構造変異の検出方法図6 がん化における変異の蓄積過程図5 全ゲノムデータ解析のフロー4 MSS技報・Vol.25変異間の距離をTSV形式で入力することでrainfall plotを標準ゲノム上に表示することができる。 全ゲノムデータ解析で得られた変異情報は、RNASeqの解析結果である遺伝子の発現量やMethylationの解析結果であるメチル化パターンなどの他の解析手法を得られる解析結果と組み合わせて総合的に確からしさを評価する。そのため、全ゲノムデータ解析の結果と他の解析手法で得られた結果を同時、かつ網羅的に確認できるゲノムビューワが必要である。GenomeJackは、NGS解析ツールが出力する主要なファイル形式をサポートしており、TSVファイル形式にも対応しているため柔軟に対応できる仕組みになっている。く、複数の変異が蓄積することにより進むプロセスであると考えられている(図6)。これとは別に、段階的に蓄積された変異だけでなく、特定の領域において一時的に変異が高頻度に発生することからがん化が進行することが分かっている[5][6]。たとえば、最近明らかになった現象としてchromothripsis、kataegis[7]と呼ばれる変異の局所的な蓄積現象があり、がん化の評価指標として用いられている。kataegisに関しては、ゲノム上に、それぞれの変異の変異間の距離(標準ゲノム上の変異間の距離)の対数をプロットするrainfall plotと呼ばれるグラフを用い、変異クラスターを検出することで構造異常を検知することができる。GenomeJackでは、図7 HER2の過剰発現5 MSS技報・Vol.253. まとめ 全ゲノム解析は網羅的にすべての変異情報を検出できることから将来有望な解析手法であることはゆるぎないが、現在のところ発展段階の解析手法といえる。全ゲノムデータ解析が発展するためには、解析の高速化、検出された変異の評価方法、検出された変異の確認ビューワなどIT技術が欠かせないものとして考えられる。 現時点においても、GenomeJackの有効性は確認できた。今後は、臨床応用機関などとの共同開発によりGenomeJackの処理の自動化と標準化を推進し、全ゲノムデータ解析を簡単に利用できる環境を構築することでゲノム診断の発展と普及に寄与したいと考えている。 最後にこれまで開発を支えてくださった方々にここでお礼申し上げたい。[1]Books,J.D(2012)Translational genomics:Thechallenge of developing cancer biomarkers,Genome Res 22:183-187[2]デヴィータがんの分子生物学, MEDSi,(2012)[3]個別化医療を拓くがんゲノム研究, 羊土社, 実験医学増刊:vol.32-No.12 2014[4]Li Ding et al, Expanding the computational toolboxfor mining cancer genomes, Nature Reviews,GENETICS, vol 15, 556-570(2014 August)[5]Stephens PJ, et al. Massive genomic rearrangementacquired in a single catastrophic event duringcancer development, Cell,144:27-40,2012[6]Campbell PJ, et al, Identification of somaticallyacquired rearrangements in cancer usinggenome-wide massively parallel paired-endsequencing, Nat Genet, 40:722-729,2008[7]Nik-Zainal S, et al, Mutational processing moldingthe genomes of 21 breast cancers, Cell, 149:979-993,2012[8]Heng Li, et al. Fast and accurate short readalignment with Burrows-Wheeler transform.Bioinformatics 2009;Volume 25, Issue 14;1754-1760.[9]Ben Lengmead et al, Fast gapped-read alignmentwith Bowtie2, Nature Methods 9, 357-359(2012)[10]Aaron Mckenna, et al. The Genome AnalysisToolkit: A MapReduce framework for analyzingnext-genneration DNA sequencing data. GenomeRes. 2010;20;1297-1303.[11]Li H et al, The Sequence alignment/map(SAM)format and SAMtools, Bioinformatics, 25, 2078-9⑶ GenomeJackを利用した事例紹介(図7、図8) 今回の事例紹介では乳がんの細胞を解析したデータを利用した(表1)(なお、本解析データには正常細胞が存在しないため、生殖細胞系列:Germlineとの比較解析は実施していない)。今回使用した解析データのサンプルは、SK-BR-3株と呼ばれるサンプルで、HER2と呼ばれる糖タンパクが過剰発現している(乳がんに対する分子標的治療薬であるトラスツズマブ(ハーセプチン®)はHER2をターゲットにしている[20])。GenomeJackを利用して、マッピング結果からペアリードの情報、クリップリードの情報から構造異常の変異を検出した。 GenomeJackは多様な表示機能(データの属性による表示色変更、プロット表示、データ値によるグラデーション表示など)、フィルタリング(データ値によるフィルタリング表示)、軽快な操作性(全ゲノムデータ解析結果を表示可能)など、がん関連変異の抽出に適した機能を有しており、これらの、がんの全ゲノムデータ解析における有効性を確認することができた。図8 chromothripsisが疑われる領域表1 検証サンプルデータ6 MSS技報・Vol.25[12]Kristian Cibulskis et al, Sensitive detection ofs o m a t i c p o i n t m u t a t i o n s i n i m p u r e a n dh e t e r o g e n e o u s c a n c e r s a m p l e s , N a t u r eBiotechnology 31, 213-219(1 March 2013)[13]Ye K, et al, Pindel:a pattern growth approach todetect break points of large deletions and mediumsize insertions from paired-end short reads.Bioinformatics, 2009 Nov 1;25(21);2865-71[14]Koboldt D., Zhang Q., et al, VarScan 2:Somaticmutation and copy number alteration discoveryin cancer by exome sequencing. Genome Res.2012;22;568-578[15]Tobias Rausch, Thomas Zichner et al. DELLY:structural variant discovery by integratedpaired-end and split-read analysis. Bioinformatics2012;28;333-339.[16]Fan X, et al, BreakDancer ‒ Identification ofGenomic Structural Variant from Paired-EndRead Mapping. Curr Protoc Bioinformatics, 2014[17]Pablo Cingolani, et al. A program for annotatingand predicting the effects of single nucleotidepolymorphisms, SnpEff: SNPs in the genome ofDrosophila melanogaster stran W1118;iso-2;iso-3. Landes Bioscience 2012;Fly6:2,1-13.[18]Wang K, Li M, Hakonarson H. ANNOVAR;Functional annotation of genetic variants fromnext-generation sequencing data. Nucleic AcidsRes. 2010;38;e164[19]Heng Li, et al. Fast and accurate short readalignment with Burrows-Wheeler transform.Bioinformatics 2009;Volume 25, Issue 14;1754-1760.[20]Slamon DJ, Leyland-Jones B, Shak S, et al. "Use ofchemotherapy plus a monoclonal antibodyagainst HER2 for metastatic breast cancer thatoverexpresses HER2." NEJM., Vol.344, No.11,2001, p.p. 783-792