テクノロジー
技術レポート:アーカイブ
Category:ライフサイエンス
ゲノムインフォマティクスの展開-大規模ゲノム配列解析から機能解明とオーミクスへ向けて
ゲノムインフォマティクスの展開を、大規模ゲノム解析、ゲノム機能解明、オーミクスの三つの領域に分け、著者らが所属していた部門が開発に携わったソフトウエアと共に概観した。大規模ゲノム解析の領域では、工程の制御・管理や効率的な情報提供を目指した工学的なソフトウエアが有効であった。ゲノム機能解明の領域では、Webベースのゲノム解析システムが機能研究において研究の進展に寄与してきた。オーミクスの領域では、複数の生物学的階層データを統合的に収納・管理するデータベースのスキーマ、時間プロファイルデータから遺伝子相互作用を解析する方法の見通しが得られた。
参考情報:
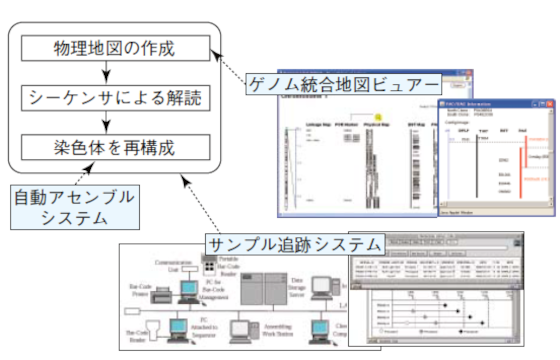
ゲノムインフォマティクスの展開-大規模ゲノム配列解析から機能解明とオーミクスへ向けて *前橋工科大学 生命情報学科 教授(博士・元つくば事業部 第三技術部) **つくば事業部 第三技術部 MSS技報・Vol.20 14技術論文ゲノムインフォマティクスの展開-大規模ゲノム配列解析から機能解明とオーミクスへ向けてExpansion of Genome Informatics : from Large-scale Genome Sequencing towards FunctionalGenome Annotation and Omics坂田克己* 清水裕司**Katsumi Sakata, Yuji Shimizu ゲノムインフォマティクスの展開を、大規模ゲノム解析、ゲノム機能解明、オーミクスの三つの領域に分け、著者らが所属していた部門が開発に携わったソフトウェアと共に概観した。大規模ゲノム解析の領域では、工程の制御・管理や効率的な情報提供を目指した工学的なソフトウェアが有効であった。ゲノム機能解明の領域では、Webベースのゲノム解析システムが機能研究において研究の進展に寄与してきた。オーミクスの領域では、複数の生物学的階層データを統合的に収納・管理するデータベースのスキーマ、時間プロファイルデータから遺伝子相互作用を解析する方法の見通しが得られた。 Here we review recent expansion of Genome Informatics by dividing it into three domains, LargescaleGenome Sequencing, Functional Genome Annotation and Omics, with special reference toparticular computer software built by authors. As for Large-scale Genome Sequencing, engineeringsoftware which aims effective process management and information-sharing was considered of value.As for Functional Genome Annotation, web-based genome analysis systems facilitated the progressof functional annotation. As for Omics, our procedures for building database schemata that integrallystore and manage the multiply-layered biological information, and for predicting gene interactionnetworks from temporal profiles, were evaluated with promising results. 1.はじめに 今世紀に入り、ヒト、イネなど我々に身近な生物種についてもゲノム配列が解読されてきた。ポストシーケンス時代とも言うべきゲノム配列解読の後段階におけるゲノムインフォマティクスの展開を整理すると、ゲノム機能解明へ向けた“情報の深化”とオーミクスへ向けた“情報の統合”の二つの方向が見えてくる。本報告では、大規模ゲノム解析、ゲノム機能解明、オーミクスの各領域におけるゲノムインフォマティクスの展開を、著者らが所属していた部門が実際に開発に携わったソフトウェアと共に概観する。本報告において紹介しているソフトウェアツールは、独立行政法人農業生物資源研究所および独立行政法人農業・食品産業技術総合研究機構・作物研究所との共同研究や開発支援を通じて三菱スペース・ソフトウエア株式会社つくば事業部(現第三技術部)のメンバーが開発に貢献したものである。2.大規模ゲノム解析におけるインフォマティクス物理地図をベースにした階層式シークエンス法によるゲノム解読の過程を図1(左上のフロー図)で示す。この方法では、BAC/PACクローン(BAC:BacterialArtificial Chromosome、PAC:P1-derived ArtificialChromosome)と呼ばれる長さ15万塩基程度のDNA断サンプル追跡システム物理地図の作成シーケンサによる解読染色体を再構成ゲノム統合地図ビュアー自動アセンブルシステム図1 物理地図をベースにした階層式シークエンス法による大規模ゲノム配列解析の流れとソフトウェアツール技術論文15片から成るライブラリを用意し、このDNA断片を主に実験的な方法により染色体上に整列化させておく。これを物理地図と呼ぶ。物理地図をベースにした階層式シークエンス法は2004年に全ゲノム解読が完了したイネゲノムプロジェクトで用いられた方法であり(1)、イネゲノムの場合には合計約4億塩基に上る12本の染色体のほぼ全体を数千本のBAC/PACクローンでカバーする。整列化したBAC/PACクローンは、長さが数千塩基程度の配列断片に分断したあと自動シーケンサーに掛けられ、A(アデニン)、C(シトシン)、G(グアニン)、T(チミン)の4種類の塩基からなる塩基配列が解読される。ここで、自動シーケンサーで一度に解読できる配列は500塩基程度に限られている。その後、自動シーケンサーにより解読された十分な数の配列断片を集め、コンセンサス配列と呼ばれる複数の断片に共通に見られる塩基配列を糊代にして、BAC/PACクローン配列を再構成する。この過程はアセンブルと呼ばれ、最適化手法の一つのダイナミックプログラミング法によりBAC/PACクローン配列の再構成が行われる。BAC/PACクローンは予め物理地図として染色体上に整列化されているので、配列が解読されたBAC/PACクローンを更に物理地図に沿って繋ぎ合わせることにより、一本が数千万塩基に及ぶ長大な染色体が再構成される。BAC/PACクローン配列の再構成をダイナミックプログラミング法に基づいて行うアセンブルエンジンは、フリーソフトウェアやCOTS(commercial off-the-shelf)ソフトウェアが出回っている。ところが、BAC/PACクローン配列の再構成にはBAC/PAC一本あたり数千個の配列断片が必要であり、更に大規模ゲノムプロジェクトでは1日に数十本のBAC/PAC配列の再構成を行う必要がある。そこで、配列断片の収集・管理、アセンブルエンジンの起動、アセンブルされた配列の管理を、誤りなくしかも効率良く行うための自動アセンブルシステムを開発した(2)。また、自動シーケンサーに掛けられる一つ一つの実験試料が各工程で誤りなく管理されていることが必要で、全体では一日当たり数万点に及ぶ膨大な試料の管理が必要であった。これに対しては、バーコードとWebシステムの応用により、実験試料がプロセスのどこを流れているか、どのくらいの作業が終了したか等の工程情報を準リアルタイムに捉えることができるサンプル追跡システムを構築した(3)。更に、イネの染色体上に整列化したマーカー配列、BAC/PACクローンなどのゲノム情報を検索・表示することができるゲノム統合地図ビュアーを開発した(http://ine.dna.affrc.go.jp/giot/GIOT.html)(4)。このビュアーでは、Javaアプレットによる動的なインタフェースを組み込んだことにより、従来の静的な画像を収納したゲノムデータベースに比し、数万個にも及ぶデータ要素が格段に高速で検索・表示できるようになった。統合マップはポジショナルクローニングにおける情報源としても活用されており、例えばイネの出穂後の止め葉や葉鞘に関するQTL(Quantitative trait locus、量的形質座位)の解析に用いられた(5)。大規模ゲノム解析では、以上のゲノムプロジェクトにおける例で見られたように、工程の制御・管理や効率的な情報提供を目指した工学的なソフトウェアが有効に機能した。3.ゲノム機能解明へ向けたインフォマティクスの展開解読されたゲノム塩基配列に対して、遺伝子として機能するゲノム領域やその遺伝子機能の予測が行われる。この作業はゲノム配列に注釈を付けることに相当し、アノテーション(Annotation)と呼ばれる。この段階では、隠れマルコフモデルに基づく遺伝子領域予測や配列類似性検索などのソフトウェアが中心的な役割を果たす。基本的なアノテーションの方法は、エキスパート研究者が主に手作業により上記ソフトウェアの結果を解釈するマニュアルアノテーションである。この時、一般に数種類~十種類程度の複数のソフトウェアを走らせ、研究者はそれらの結果を解釈して適当な結果を抽出し、注釈データとして記録するという事を行う。これらアノテーション作業を効率化するため、(i)アノテーションの材料となる複数のソフトウェアの解析結果を一括して提供することにより、高速かつ標準的なマニュアルアノテーションを促す、(ii)一般のWebユーザに対しマニュアルアノテーションを近似したアルゴリズムに基づく自動アノテーションのメカニズムを提供する、を目標にイネゲノム自動アノテーションシステムRiceGAAS(6)(http://r i c e g a a s . d n a . a f f r c . g o . j p / ) を開発した。図2 はRiceGAASによるアノテーションの概念図である。箱形atacacccacatataggcacatgctaaacttctacactggaagctagcatatatccattc…図2 ゲノムアノテーションの概念(グラフィック出力はRiceGAAS(http://ricegaas.dna.affrc.go.jp/)によった)MSS技報・Vol.20 16や矢印形のシンボルが予測されたゲノム機能領域を示している。緑色の矢印が予測遺伝子領域を表している。矢印の向きが図の上段と下段で異なるのは、互いに相補的配列を持つ二本のDNA鎖それぞれにおける予測遺伝子を示しているからである。RiceGAASは、遺伝子領域予測、配列類似性検索などの15種類に及ぶ各種解析プログラムを組み込み、それらの結果をある種の多数決処理で解釈し、ゲノム配列上に候補遺伝子領域を予測している。上記アノテーションシステムは2002年に公開した後も、様々な改良に取り組んでいる。その一つはCpGクラスタ(CG塩基の並びが特異的に見られる領域)と遺伝子の位置関係を解析する機能の組み込みである(http://ricegaas.dna.affrc.go.jp/CpG/)。この機能は、イネの染色体上におけるCpGクラスタと遺伝子の位置関係に特徴的なパターンを見つけるのに使われた(例えば、遺伝子の一端にCpGクラスタを持つ遺伝子は染色体上にランダムに位置するのではなく、遺伝子群として固まって位置しているといった特徴)。尚、上記のような位置関係は脊椎動物において知られていたが、それがイネゲノム上でも発見されたことで、脊椎動物と同様の遺伝子発現機構が植物においても示唆された(7)。また、イネの予測遺伝子モデルに対し、配列の類似性を基準に遺伝子機能の予測と遺伝子の分類を行うGFSelector(http://alnilam.mi.mss.co.jp/rgadb/)を開発してRiceGAASに組み込んだ。このGFSelectorとRiceGAASからなる統合システムを、コムギ、バナナ、ダイズのゲノム配列に試験的に適用したところ、イネゲノムに適用した場合と同程度の予測精度が得られることが示された(8)。更に上記アノテーションシステムは、特定の機能を持つ遺伝子の同定、例えばイネ収量調節遺伝子の解析(9)、着色米が生じるための酵素遺伝子と調整遺伝子の解析(10)、に用いられてきた。RiceGAASは30カ国以上で利用実績があり、現在も月当たり数千件のアクセスが継続的にある(図3)。特に国内からのアクセスよりも、国外からのアクセスの方が多いのが特徴である。これは、RiceGAASが海外において、イネ科作物やバナナなどイネ以外の単子葉植物のゲノム解析に利用されている為であると推察される。また、RiceGAASはカイコゲノム自動アノテーションシステムK a i k o G A A S ( h t t p : / /kaikogaas.dna.affrc.go.jp/)の基盤にもなっている。RiceGAASはイネゲノム解析を目標に開発されたシステムであったが、機能研究へ向けた深化や近縁種におけるゲノム解析の展開に伴って、長い期間に亘り幅広く利用されてきた。今後もRiceGAASのような利用者のニーズに沿ったアプリケーションシステムの開発が重要になるであろう。本章では、Webベースの自動アノテーションシステムが、ゲノム機能研究においてもツールとして受け入れられ、研究の進展に寄与してきたことを示した。従来から、解読される配列量に対し手作業でアノテーションされる配列量が追い付かないという指摘がなされているが(11)、この差異は桁違いに速い次世代型シーケンサーの普及により益々増大することが懸念される。我々は、自動アノテーションシステムがこの問題の解決の一助になると考えており、引き続き精度向上などの改良を図っていく所存である。4.オーミクスへ向けたインフォマティクスの展開ゲノムに引き続き、転写産物(トランスクリプトーム)、タンパク質(プロテオーム)や代謝産物(メタボローム)など様々な生物学的階層における情報の収集・解析が行われるようになってきた。図4は生物学的階層データ統合の概念を示す。様々な生物学的階層の情報を組み合わせ、ネットワーク上に位置付け、システムとして生命現象の解明を目指すことが益々重要になると予想される。我々は複数の生物学的階層に跨るデータベースとしてダイズプロテオームデータベース(http://proteome.dc.図3 イネゲノム自動アノテーションシステムのアクセス履歴(http://www.dna.affrc.go.jp/statistics/RiceGAASt.html) 図4 生物学的階層データ統合の概念メタボロームゲノムトランスクリプトームプロテオーム技術論文17affrc.go.jp/Soybean/)を開発した(図5)。当データベースにおいて主となるプロテオームデータは、タンパク質二次元電気泳動に基づくデータであり、出芽後1週間の水ストレスを与えたダイズの各部位および細胞内小器官で検出された約7300タンパク質スポットを収納している(そのうち約500タンパク質が同定されている)。データベースにはトランスクリプトーム(転写産物)およびメタボローム(代謝物)のデータも収納し、異なる生物学的階層間のデータ関連付けを行った。更に、各階層の時間プロファイルデータを統合的に解析した結果から、水ストレスにより特定の代謝系で代謝量の上方調節が引き起こされる等が示唆された(12)。本データベースでは収納している時間プロファイルデータの一つ一つに対し、プロファイルの特徴(上昇、下降および極値の存在)を簡潔に示すタグ情報を付加した。これにより、ユーザは時間プロファイルの特徴に基づいた情報検索を行うことができるようになった。また、トランスクリプトーム、プロテオーム、メタボロームの各層に共通したデータ形式の一つが時間プロファイルデータである。時間プロファイルデータは遺伝子やタンパク質の発現量、あるいは代謝産物量の時間変化を示すデータである。このような時間プロファイルデータから遺伝子間の相互作用を有向グラフとして推定する方法を開発し( 13)、MINOS( Mathematical geneInteraction Network Optimization Software)というプログラムに実装した。この推定法は、Sシステム微分方程式の近似式に基づいた高速アルゴリズムであり、100個程度の遺伝子からなるネットワークについても容易に解析できるようになった。図6は、時間プロファイルデータとSシステム微分方程式に基づく遺伝子相互作用ネットワーク推定の概念を示す。MINOSの適用例として、イネの幼苗期の生長に伴うタンパク質の発現解析と機能解析がある(14)。また、ダイズの出芽期において水ストレスに応答するタンパク質群を解析する研究にも用いられた(15)。上記の研究では、タンパク質の時間プロファイルデータから、それぞれ61個(14)および51個(15)のタンパク質間の相互作用ネットワークを推定した。上記のようにオーミクスへ向けた展開では、複数の生物学的階層のデータを統合的に収納・管理するデータベースのスキーマ、時間プロファイルデータから遺伝子相互作用を解析する方法が生物学的機能の解明に役立つ見通しが得られた。5.むすび大規模ゲノム解析およびポストシーケンス時代におけるゲノムインフォマティクスの展開を著者が所属した部門が開発に携わったソフトウェアと共に概観した。大規模ゲノム解析ではプロセス制御・管理や効率的な情報提供を行う工学的なソフトウェアが有効であった。ゲノム機能解析では、Webベースの解析システムがゲノム機能研究においても受け入れられ、研究の進展に寄与していた。オーミクスの領域では、複数の生物学的階層におけるデータを統合的に収納・管理するデータベースのスキーマ、時間プロファイルデータから遺伝子相互作用を解析する方法の見通しが得られた。今後はオーミクスとゲノム機能解析の二つの領域に加え、次世代型シーケンサーの動向にも注視して、更なる技術力向上を図っていく必要があると考える。謝辞本報告で紹介しているソフトウェアツールの開発に参加した三菱スペース・ソフトウエア株式会社つくば事業部(現第三技術部)メンバー、および本原稿のレビューを引き受けて戴いた、長村吉晃博士、松本隆博士(独立行政法人農業生物資源研究所所属)、小松節子博士(独立行政法人農業・食品産業技術総合研究機構・作物研究所所属)に感謝いたします。図5 生物学的階層データを統合したデータベースの例(ダイズプロテオームデータベース、http://proteome.dc.affrc.go.jp/Soybean/)時間時間時間発現量x1x2xn発現量の時系列に注目生成項分解項Sシステムモデル(質量作用則に基づく 微分方程式モデル)図6 時間プロファイルデータとSシステムモデルに基づく遺伝子相互作用ネットワーク推定の概念MSS技報・Vol.20 18参考文献盧International Rice Genome Sequencing Project:The map-based sequence of the rice genome.Nature, 436, 793-800(2005)盪Antonio,B.A., Nobushima S., Honda S., KanamoriH., Yamagata H., Yamamoto K., Matsumoto,T.,Sasaki,T., Sakata,K.: An Auto-Assembly Systemfor Rice Genome Sequencing. 5th AnnualConference on Computational Genomics, P-01,pp.15(2001)蘯Sakata K., Waki K., Sasaki T., Simomura M., KiseM.: A Sample Tracking Tool for Rice GenomeSequencing. Genome Informatics, 9, 222-223(1998)盻Sakata K., Antonio B.A., Mukai Y., Nagasaki H.,Sakai Y., Makino K., Sasaki T.: INE: a RiceGenome Database with an Integrated Map View.Nucleic Acids Res., 28, 97-101(2000)眈Kanbe T., Sasaki H., Aoki N., Yamagishi T., Ohsugi R.: The QTL Analysis of RuBisCO in FlagLeaves and Non-Structural Carbohydrates in LeafSheaths of Rice Using Chromosome SegmentSubstitution Lines and Backcross Progeny F-2Populations. Plant Production Science, 12, 224-232(2009)眇Sakata K., Nagamura Y., Numa H., Antonio B.A.,Nagasaki H., Idonuma A., Watanabe W., ShimizuY., Horiuchi I., Matsumoto T., Sasaki T., Higo K.:RiceGAAS: an Automated Annotation System andDatabase for Rice Genome Sequence. NucleicAcids Res., 30, 98-102(2002)眄Ashikawa I., Numa H., Sakata K.: SegmentalDistribution of Genes Harboring a CpG Island-LikeRegion on Rice Chromosomes. Molecular Geneticsand Genomics, 275, 18-25(2006)眩Sakata K., Ikawa H., Watanabe H., Ashikawa I.,Shimizu Y., Horiuchi I., Antonio B.A., Numa H.,Nagamura Y., Matsumoto T.: ・A BioinformaticsResource for Crop Functional Genomics:GFSelector Module in Automated AnnotationSystem, RiceGAAS. JARQ, 43, 103-113(2009)眤Ashikari M., Sakakibara H., Lin S., Yamamoto T.,Takashi T., Nishimura A., Angeles E.R., Qian Q.,Kitano H., Matsuoka M.: Cytokinin OxidaseRegulates Rice Grain Production. Science, 309, 741-745(2005)眞Furukawa T., Maekawa M., Oki T., Suda I., Iida S.,Shimada H., Takamure I., Kadowaki K.: The Rcand Rd genes are involved in proanthocyanidinsynthesis in rice pericarp. Plant Journal, 49, 91-102(2007)眥Maier D.: Automatic protein function predictionusing the Pedant-Pro expert system. ISMB/ECCB(2007)眦Sakata K., Ohyanagi H., Nobori H., Nakamura T.,Hashiguchi A., Nanjo Y., Mikami Y., Yunokawa H.,Komatsu S.: Soybean Proteome Database: A DataResource for Plant Differential Omics. J. ProteomeRes., 8, 3539_3548(2009)眛Mitsui S., Nobori H., Takada T., Kishida K., MiuraY., Ikawa H.: Development of a New Gene-Network Estimation System. Genome Informatics,14, 384-385(2003)眷Tanaka N., Mitsui S., Nobori H., Yanagi K.,Komatsu S.: Expression and function of proteinsduring development of the basal region in riceseedlings. Molecular & Cellular Proteomics, 4, 796-808(2005)眸Hashiguchi A., Sakata K., Komatsu S.: ProteomeAnalysis of Early-Stage Soybean Seedlings underFlooding Stress. J. Proteome Res., 8, 2058_2069(2009)